Neuroblastoma
Anatomy
Epidemiology
Neuroblastoma is the most common extra-cranial pediatric malignancy and accounts for 15% fo cancer related deaths. It arises from embryonic neural crest cells of the peripheral sympathetic nervous system. There is marked clinical heterogeneity, leading to differences in behavior from spontaneous maturation to rapid proliferation and metastatic progression.
There are about 650 cases in the US/year at 10.3/million from birth through 15 years. It is the most comon malignancy in infants (≤ 18 months). 90% are diagnosed by 10 years, and 79% by 4 years. The median age at diagnosis is 22 months.
Pathology
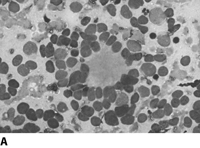
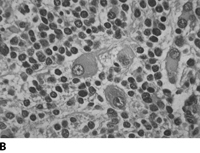
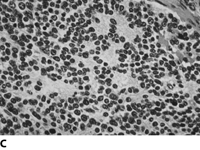
Neuroblastoma is a small round blue cell tumor. Homer-Write pseudorosettes are found in 15-50% of the cases. They are classified by the Shimada Classification into favorable and unfavorable histology which is based on age, amount of Schwann cell stroma, nodular v. diffuse pattern, degree of differentiation and mitotic index.
 |
 |
 |
The original neuroblastoma histology classification was the Shimada classification. Shimada was a "favorable v. unfavorable" grading independent of N-myc amplification. It was based on stroma, age, differentiation adn mitotic index. Favorable Shimada was stroma rich, all ages, no nodular pattern, or stroma poor, age 1.5-5 years, differentiated, mitotic index < 100 or stroma poor, age < 18 months, mitotic index < 200. Unfavorable is age > 5, stroma poor, or stroma rich, all ages with nodular pattern.
The present INRG histology staging keeps much of Shimada intact, but subcategorizes and renames some categories.
| International Neuroblastoma | Pathology Classification | Original Shimada Classification | Prognostic Group |
|---|---|---|---|
| Neuroblastoma Favorable | (Schwannian stroma-poor)aFavorable | Stroma-poor | Favorable |
| <1.5 years | Poorly differentiated or differentiating and low or intermediate MKI tumor | ||
| 1.5–5 years | Differentiating and low MKI tumor | ||
| Unfavorable | Unfavorable | Unfavorable | |
| <5 years | (a) Undifferentiated tumorb >(b) High MKI tumor |
||
| 1.5–5 years | (a) Undifferentiated or poorly differentiated tumor
>(b) Intermediate or high MKI tumor |
||
| ≥5 years | All tumors | ||
| Ganglioneuroblastoma, intermixed | (Schwannian stroma-rich) | Stroma-rich intermixed (favorable) | Favorablec |
| Ganglioneuroma | (Schwannian stroma-dominant) | ||
| Maturing | Well differentiated (favorable) | Favorablec | |
| Mature | Ganglioneuroma | ||
| Ganglioneuroblastoma, nodular | (Composite Schwannian stroma-rich/stroma-dominant and stroma-poor) | Stroma-rich nodular (unfavorable) | Unfavorablec |
| Stage | Elements |
|---|---|
| 1 | Localized tumor with GTR ±microscopic residual. Adherent LN may be positive, but non-adherent nodes must be negative |
| 2A | Localized tumor with < GTR, ipsilateral non-adherent LN negative. | 2B | Localized tumor with ipsilateral nonadherent LN+, contralateral LN negative |
| 3 | Unresectable tumor; tumor extends across the midline (defined as opposite side vertebral body), contralateral LN negative |
| 4 | Mets to distant LN, bone, bone marrow, liver, skin or other organs |
| 4S | Age < 1 year, otherwise Stage 1-2B primary with metastasis limited to skin, liver and/or < 10% of bone marrow (MIBG scan should be negative) |
Cytogenetic abnormalities are associated with poor prognosis: n-myc amplification, diploid tumors and increased telomerase activity.
| COG Risk groups based on INSS Stage |
|---|
| Low Risk (OS3 > 90%) |
| Any Stage I |
| Stage 1 and age < 1 year |
| Stage 2 and age > 1 year without N-myc ampl |
| Stage 2 and age > 1 year with N-myc ampl, FH and hyperdiploid |
| Stage 4S without N-myc amplified, FH, hyperdiploid |
| COG Risk groups based on INSS Stage |
|---|
| Intermediate Risk (OS3 70-90%) |
| Stage 3 and age < 1 year without N-myc amplified |
| Stage 3 and age > 1 year without N-myc ampl. and FH |
| Stage 4 and age < 18 months without N-myc ampl. |
| Stage 4S and age < 12 months without N-myc with UH or diploid |
| COG Risk groups based on INSS Stage |
|---|
| High Risk (OS3 30%) |
| Stage 2 and age > 1 year with N-myc amplified and UH |
| Stage 3 and age < 1 year with N-myc ampl |
| Stage 3 and age > 1 year with N-myc ampl, UH or diploid |
| Stage 4 and age < 18 months with N-myc ampl |
| Stage 4 and age > 18 months |
| Stage 4S and age < 1 year with N-myc ampl. |
Natural History
Neuroblastomas are frequently found in the adrenals. Neuroblastoma nodules have been found in situ in adrenal glands at autopsy from infants who have died of other causes, suggesting that most of these nodules regress by birth. 90% secrete VMA and HVA and can be detected by analyzing urinary catecholamines.
Staging Systems
The INSS was developed to try to provide a uniform staging system so studies could be compared. It is an attempt at merging prior CCG and POG staging systems. It is a surgical staging system and is limited, particularly in Stage 3, in that surgery is not always done at diagnosis and many Stage 2 patients do not get extensive LN sampling. As a result, the INRGSS (international neuroblastoma research group Staging System) developed yet another staging system based on imaging and the presence or absence of 1 or more of 20 imag-defined risk factors.
Non-metastatic disease is characterized as L1 or L2, while metastatic disease is characterized as M and MS. In MS, disease is confined ot he skin, liver or bone marrow in children < 18 months old.
| Stage | Description |
|---|---|
| L1 | Localized tumor not involving vital structures as defined by the list of image-defined risk factors and confined to one body compartment |
| L2 | Locoregional tumor with presence of one or more image-defined risk factors |
| M | Distant metastatic disease (except stage MS) |
| MS | Metastatic disease in children younger than 18 months with metastases confined to skin, liver, and/or bone marrow (<10%) |
| Monclair T, Brodeur GM, Ambros PF, et al. The International Neuroblastoma Risk Group (INRG) staging system: an INRG task force report. J Clin Oncol. 2009;27(2):298–303 | |
The imaging defined risk factors are described succinctly by M. B. McCarville available here: Imaging neuroblastoma: what the radiologist needs to know. Cancer Imaging. 2011 Oct 3;11 Spec No A:S44-7.
Clinical Workup and Evaluation
Physical and History
The general clinical workup includes looking for classic signs of neuroblastoma: "blueberry muffin" sign consisting of blue non-tender skin nodules, raccoon eyes suggestive of orbital metastases with proptosis and bruising, opsoclonus-myoclonus-truncal ataxia (as a result of paraneoplastic syndrome = myoclonic jerking and random eye movements that may persist after cure).
These kids look sick. An ill child with an abdominal mass is more likely to be a neuroblastoma than a Wilms tumor.
Labs
Laboratory studies include standard blood and chemistries, but also urinary catecholamines: VMA, HMA.
Imaging
Imaging includes CT/MRI of primary site, MIBG scan and CXR. If chest x-ray is positive, then order a chest CT. There are calcifications seen on x-ray in the primary in 80%-90% of the cases. If the primary tumor does not enhance on MIBG scan, then get a bone scan.
Tissue Diagnosis
The primary or involved nodes should be biopsied. All patients should have a bilateral bone marrow biopsy.
General Management and Treatment
Low Risk Disease
Low risk tumors are managed with surgery alone unless there is impending cord compression, or respiratory compromise. A brief course of chemotherapy may be required in these situations. Low risk stage 1/2 disease have an OS4 > 95% with surgery alone. Stage 1/2 disease is resectable. For infants that have Stage 4S disease, survival is also > 90% with supportive care or a short course of chemotherapy.
Intermediate Risk Disease
Intermediate risk disease consists of infants and toddlers without N-myc amplification. These patients are expected to have an 80% survival rate with moderate doses of chemotherapy for 4-8 months and primary tumor resection. Radiation is rarely used in the low or intermediate risk groups because of the favorable prognosis and the young age of the patients.
High Risk Disease
The high risk group is primarily Stage 4 and older than 1 year at diagnosis. It also includes Stage 3 with N-myc amplified or unfavorble histology. The OS for children older than 18 months with high risk disease has increased from about 15% to 30-40% now, with increasing use of myeloablative therapy.
Treatment for high risk disease is divided into 3 phases: intensive induction therapy (chemotherapy), marrow ablative therapy, and management of the minimal residual disease. The goal of the induction regimen is to maximally reduce tumor burden, including bone marrow burden within a time frame that minimizes the risk of development of resistant clones. At the end of the induction, local control of bulky disease is accomplished either before or after myeloablative therapy with either surgery or radiation. Then high dose marrow ablative therapy may be used to try to overcome residual disease followed by stem cell transplant.
Chemotherapy
Chemotherapy is used in low risk disease only for recurrence or for compromised organ function. In intermediate risk disease, long term survival is acheived with moderate combination chemotherapy for 4-6 months.
The most effective chemotherapy regimens for induction are combinations of platinum based, cyclophosphamide, doxorubicin, etoposide, vincristine and ifosfamide. Induction regimens have a 60% -80% overall response rate at the end of 5-6 months of treatment. Most recently COG has added topotecan with cyclophosphamide. EU is testing a dose-intensive schedule. The rapid group (dose-intensified) appears to have improved EFS5 but did not change OS.
Radiation
A key goal is to cure neuroblastoma while avoiding potentially life long sequelae of treatment. In vitro nb is radiosensitive, but in vivo, the response is less predictable. Prior to chemotherapy radiation was used successfully in Stage 2 and 3 disease with 100% survival. But staging systems, histology are all different and have evolved over time.
For low risk disease, PORT is unnecessary in Stage 1/2 disease. All recent cooperative group studies have reported more than 90% survival with surgery alone or surgery with chemotherapy. GTR cures the majority regardless of age, VMA/HVA ratio, or histology. Radiation is reserved for the very rare situation where local recurrence or persistance of disease exists, despite surgery and chemotherapy and when function is threatened, such as spinal cord compression or respiratory insufficiency caused by hepatosplenomegally.
For Stage 4S, hepatosplenomegaly threatening respiratory compromise, RT can be used for palliation, balanced against the need for risk of late sequelae. This occurs most commonly in 1-2 month olds, which is a risk factor for death through IVC obstruction, renal perfusion compromise, GI compromise or DIC. Low dose chemotherapy can be attempted, but often in very young infants, the progression is too rapid and local radiation is indicated for acute liver. 2 Gy - 6 Gy is usually adequate. (1.5 Gy x 3 fractions = 4.5 Gy)
For intermediate risk disease, several reports discuss a survival advantage with RT. Patients with Stage 3 (inoperable) who are > 1 year old, with N-myc amplification or unfavorable histology need further treatment. These children are expected to have an EFS of 50-60%, even with aggressive multi-mode therapy.
POG did a randomized study to test radiation with patients > 1 year old, Stage C disease → chemotherapy ±radiation. RT dose was 24-30 Gy in 16-20 fractions at 1.5 Gy/fraction. Radiation increased the CR/DFS from 45% to 67%. All current POG/COG protocols prescribe radiation after chemotherapy for residual disease, with the exception of infants and children with favorable biology (defined as without N-myc amp and hyperdiploid).
For HIGH risk disease(about half the cases) prognosis remains poor. Less than 30% older than 1 year old survive more than 5 years. Early large field radiation created significant toxicity. Aggressive chemotherapy, and stem cell transplant with TBI as a component have been used. An enlarging body of evidence for RT to local sites including the primary site and residual bulky metastatic disease may be advantageous.
Radiation Therapy Treatment Planning And Techniques
Volume
RT volumes are determined by the imaging studies and the surgeon's operative note and clip placement. If LN involvement is suspect or demonstrated, a wide field that covers the primary tumor site and nodal drainage is appropriate. If the field must cover a portion of the vertebral body, the entire VB shall be covered to avoid scoliosis. There is controversy regarding the need to cover second echelon LN. Relapse in on-iradiated second echelon nodes does occur ,but usually in conjunction with local or distant mets. Few cases involve isolated second echelon failure and including them creates a greater source of late sequelae. Care should be taken to avoid to the maximum extent feasible organs at risk and normal uninvolved tissue. OAR tissues may be much more sensitive in infants therefore, doses should be avoided to these organs, if at all possible.
- GTV
- Post-induction chemotherapy volume but volume prior to surgery.
- CTV
- Unspecified, but I think 1.0 cm is reasonable. Treat this field to 21.6 Gy
- Boost CTV
- Any gross residual post-op disease after induction chemotherapy and surgery. Treat this volume to 14.4 Gy (total dose 36 Gy).
- CTV-mets
- Treat any mets still positive on MIBG/PET after induction chemotherapy to 21.6 Gy.
Dose
Due to apparent limited NB repair ability, and some NB tumors have a rapid doubling time, shorter courses of radiation, including BID and hyperfractionation may be advantageous. The overall treatment elapsed time may also be significant and radiation course should be kept short. Clinical response is variable. In-field recurrences are rare, but do occur. Based on CCG 3891 (mediastinal and abdominal sites) treated tumors to 10 Gy to the primary, but a subgroup received 20 Gy to extra-abdominal tumors. Local relapse rates were 0% (with EBRT) and 44% (no radiation).
A basic guide for patients with high risk neuroblastoma recommends a minimum dose of 21 Gy at 1.8 Gy/fraction or 1.5 Gy BID should be delivered to the pre-operative/post-chemotherapy tumor volume and regional lymphatics.
The ANBL0532 study for patients who have an incomplete resection prescribes 21.6 Gy EBRT to the post-induction chemotherapy, pre-operative tumor volume → boost 14.4 Gy (36 Gy total) to any gross residual diseae volume. A Köln, Germany report suggested that 36 Gy could compensate for the disadvantage of incomplete response to chemotherapy. They reported improved outcome following EBRT that EFS3 100% OS3 100% compared to no radiation, despite residual tumor.
Outcomes, Patterns of Failure, Prognostic Indicators
Several clinical and biologic factors are associated with prognosis. As neuroblastoma has a highly variable clinical course, and neo-natal screening did not change outcomes, other than to lead to more extensive treatment, prognostic indicators become more important to avoid treating disease that could resolve on its own, while insuring treatment for disease that could very well be lethal if left untreated.
INRG showed that patients could be stratified into 4 different risk groups for EFS5 using:
- age
- Stage
- histology
- grade of differentiation
- N-myc
- ploidy
- 11q aberration
Age and stage at diagnosis are extremely important determinants. The stage strongly influences prognosis. With surgery as a primary treatment, Stage 1/2 have a 90% survival rate, and State 4S has an 85% survival rates. With multi-modality therapy, State 3 survival rates are 60%-90%. Survival drops to 40% for stage 4.
Age is a key prognostic indicator with younger infants doing better than older infants. Presently, the age cutoff for favorable outcomes is ≤ 18 months, if the biology is favorable. Infants under 18 months do better than older children of the same stage.
Initial response to treatment is a favorable prognostic factor. INRG has published a standardized response chart ranging to very good response through progressive disease. An early response by MIBF scan after 2 -4 cycles of induction chemotherapy for Stage 4 disease has been shown to correlate with event free survival.