Other Gliomas
Optic Path Gliomas
These account for about 5% of pediatric tumors. The peak is in the very young at 2-6 years and 75% are younger than 10. 1/3 have neurofibromatosis. These tumors are grouped into:
- tumors confined to the optic nerve
- tumors of the optic chiasm (anterior tumors)
- tumors involving the hypothalamus or adjacent structures (posterior tumors)
Bilateral Optic Nerve Involvement = NF-1 = NO RADIATION!
Bilateral optic nerve (ON) involvement is generally considered pathognomic for NF-1 and are often incidentally found on routine imaging. The frequency of progression is uncertain with estimates from < 10% to 40-50% in series reported by oncology centers. Even in symptomatic patients, only 30-50% will progress to need treatment. The initial management is close observation with regular opthalmologic exams and MRI. On progression or increasing symptoms, chemotherapy is the initial treatment, reserved for those with clear evidence of progression causing increased symptoms. NF-1 and radiation to the brain do not get along very well with each other!
Unilateral Optic Nerve Involvement
Unilateral optic nerve involvement may not be related to NF-1. These patients present most frequently with proptosis which may be long-standing. Findings of optic atrophy and impaired visual acuity are seen. MRI imaging demonstrates small well circumscribed lesions with bright enhancement typical of pilocytic astrocytoma. Biopsy is not necessary for diagnosis. Treatment will usually be necessary, with the approach depending on the extent of useful vision remaining.
Surgery is used if there is no useful vision remaining, and consists of enucleation and resection of tumor.
If vision is preserved, chemotherapy is the first line treatment in infants and children up to age 5, and for ALL patients with NF-1. Radiation therapy can be considered for older patients. Overall the prognosis is good and visual acuity remains stable or improves in the majority following treatment with either chemotherapy or radiation therapy. Long term tumor control approaches 100% with either treatment.
Optic Chiasm Gliomas
These tumors involve the chiasm or hypothalamus. These tumors appear as well circumscribed lesions on MRI with uniform bright enhancement. Patients present with visual field cuts (temporal fields) and loss of acuity. Biopsy is not usually necessary. These tumors should be observed as initial management, particularly for NF-1 patients. For younger than 5 year old patients particularly where NF-1 is involved, there is high probability of early progression and these patients will usually require early treatment within a few months after diagnosis. Surgery is rarely a viable option for tumors in this location. Chemotherapy is usually the initial treatment of choice for infants and young children and patients with NF-1. Radiation therapy is used for salvage after chemotherapy and for definitive treatment of older children without NF-1, provided there is reasonable expectation that vision will not further deteriorate.
The overall survival for patients with optic chiasm tumors is 90% - 100%. The probability of long term PFS is 60% - 90% range.
Posterior/Chiasm/Hypothalamic gliomas
These tumors account for 70% of optic path gliomas in children, are typically rather large, lesions that are thought to arise in the chiasm and extend to the hypothalamus. They can extend posteriorly along the optic tracts and often fill the 3rd ventricle, causing hydrocephalus. Early findings consist of nystagmus, impaired vision, with late presentations include increasing head circumference and signs of increased ICP. Treatment consists of CSF diversion to relieve ICP usually through a VP shunt and if necessary, surgical resection, particularly if the tumor is exophytic and growing into the basal cistern. Surgery in these circumstances provides rapid relief of symptoms. The resection will be incomplete. Surveillance following surgery is reasonable although most will eventually progress, requiring additional treatment.
PFS at 3-5 years is 20% to 60%, at best. Some patients will not have progression. For those that do progress, chemotherapy will delay the need for radiation for a median of 2 - ≥ 4 years without jeopardizing OS.
Indications for radiation therapy are:
- progression on chemotherapy for kids younger than 10
- progression of disease at diagnosis or after surgery for older patients.
Radiation therapy is a local treatment with doses of 45 Gy - 50 Gy for younger children and 50 Gy - 54 Gy for children ≥ 5 years old.
Patients with NF-1 are at risk for severe radiation sequelae. They may have subnormal IQ even without chemotherapy ± radiation. They are also at risk for developing moya moya syndrome. MRI-angiography is useful in regular follow up to insure patent circle of willis which is the underlying pathology of Moya Moya to prevent a stroke.
Brainstem Gliomas
Tumors arising in the pons, mid-brain and medulla account for 10% - 15% of all pediatric CNS tumors. These are classified into distinct types:
- favorable low grade focal disease
- dorsal exophytic
- cervicomedullary tumors
- Diffuse intrinsic pontine tumors (aggressive)
Focal tumors are limited in size (< 2 cm), well circumscribed on MRI without evidence of infiltration and without edema. They may be cyustic and the cystic component may be large relative to the solid component. Focal tumors can occur at any level of the brainstem, but most frequently occur in the midbrain and medulla. They usually have a long history of localizing findings such as an isolated cranial nerve deficit and contra-lateral hemiparesis. Increased ICP is uncommon, unless the tumor is in the tectal plate that may stenose the aquaduct of Sylvius while still small.
Management depends on the location of the tumor within the brainstem, and the imaging characteristics. Pagients with non-enhancing focla tumors in the tectal region may do well with only procedures to relieve ICP such as ventriculostomy. Active intervention, including biopsy, is reserved for patients with clinical and radiologic evidence of tumor progression. Surgery in this area is frought with the substantial risk of morbidity even with modern techniques. Surgery is, however, the technique of choice for exophytic tumors that are accessable and have characteristics of low grade histology.
For inoperable tumors, conventional radiation therapy in brainstem tumors correlates with location: pons v. elsewhere, imaging findings (volume, density, enhancement pattern), and histology. About 50% to 70% may be controlled permanently.
Standard Treatment is as for low grade gliomas. External beam, conventionally fractionated radiation at 1.8 Gy/fraction to a total dose of 54 Gy. The treatment volume is the CTV= GTV + 0.5 cm and the PTV should include the institutional setup uncertainty.
Surgery is the treatment of choice for dorsal exophytic tumors, using intra-operative image guidance to achieve a maximum safe resection. Due to the intimate involvement with the brainstem surface, some tumor is residual, but children do well and post-operative adjuvant treatment is not routinely recommended. Radiation may be indicated when the tumor is found to be high grade or for those who develop early progression (< 9 months post-op).
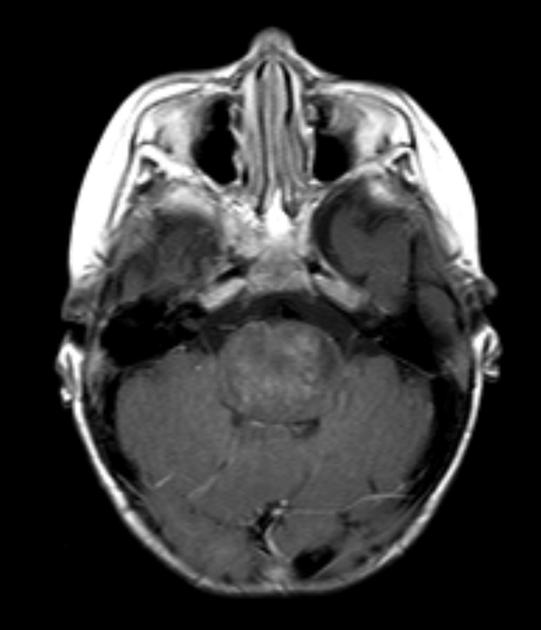
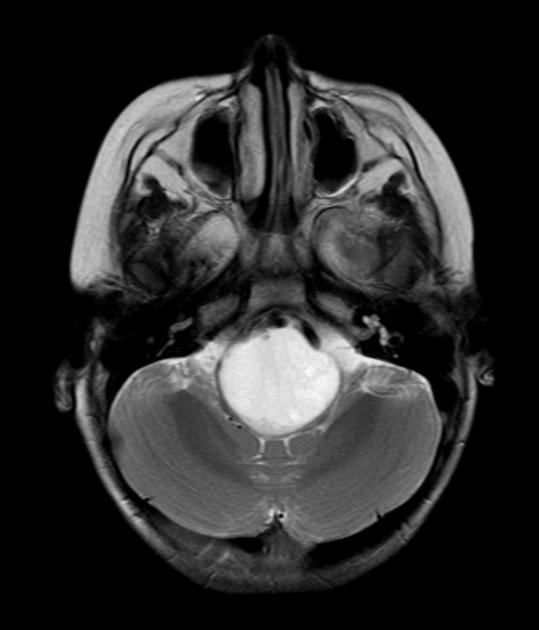
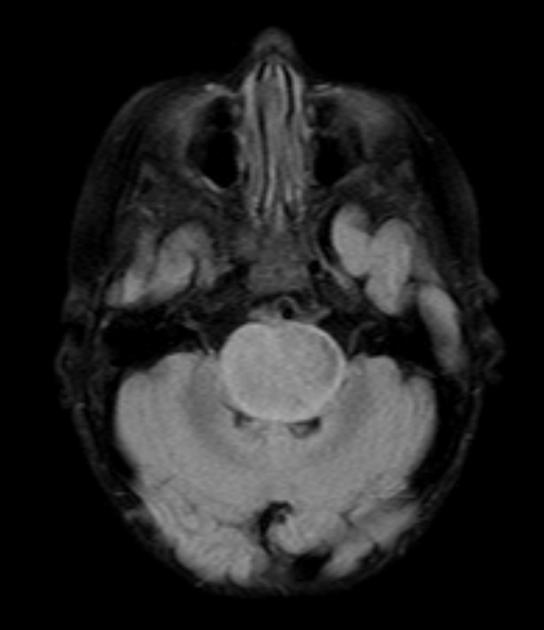
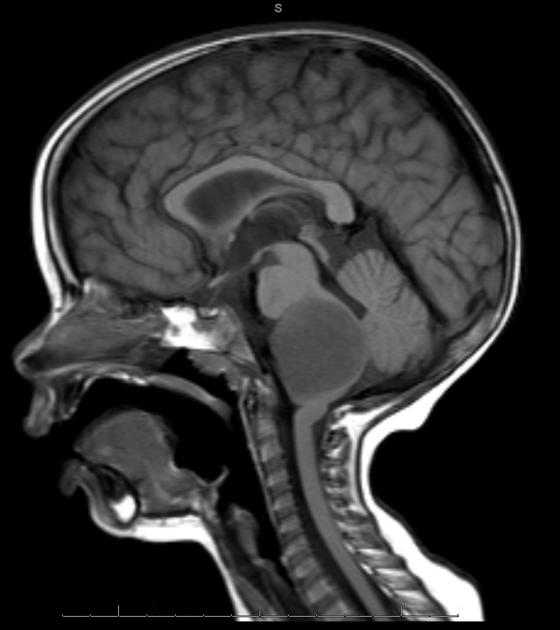
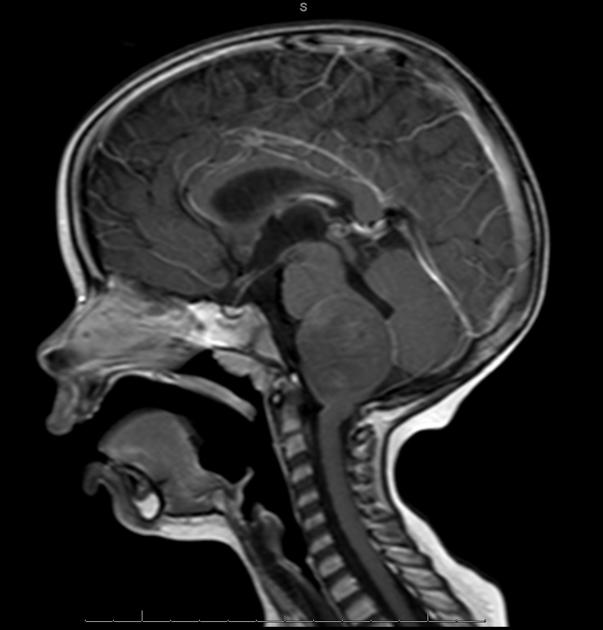
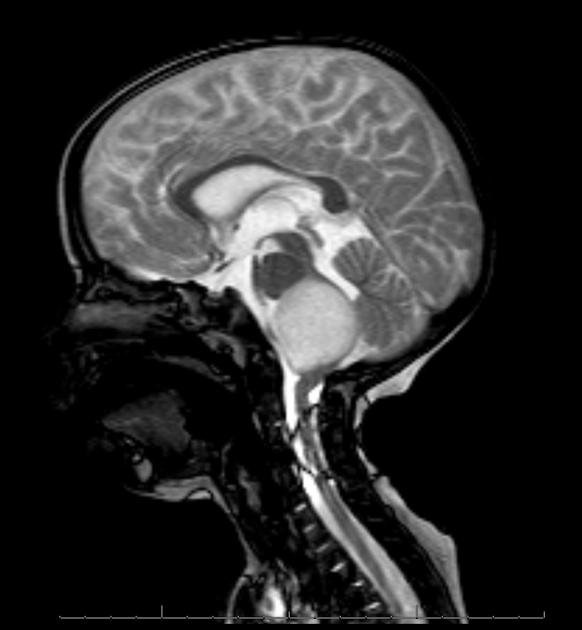
Diffuse Pontine Glioma
These tumors account for 70% to 80% of all brainstem tumors and they are deadly. They arise in the pons, and cause diffuse enlargement of the brainstem. They extend to the midbrain and medulla. Exophytic growth is seen in at least 2/3 of the cases. The majority of these are fibrillary astrocytomas with a propensity for malignant transformation. They carry a very poor prognosis.
They present with a very short onset and duration of symptoms:
- multiple bilateral cranial nerve deficits, especially VI and VII
- Long tract signs
- ataxia
- 10% have hydrocephalus at diagnosis
They appear on MRI T2 or FLAIR. The presence of ring enhancement suggests high grade disease. See Radiopedia for more images of diffuse pontine gliomas.
|
|
|
|
|
|
Surgery has no role in the treatment of diffuse pontine gliomas. It is dangerous to biopsy them, the imaging and physical findings are characteristic and histology does not change management.
Radiation therapy treatment will lead to improvements in the majority (70%) of patients, but the results are transient with a PFS of < 6 months and poor survival with a median survival of < 1 year and an OS2 of < 20%. All attempts to improve this have been futile.HFRT was tested with dose escalation up to 78 Gy. Time to progression and overall survival were not improved over conventional treatment and morbidity was considerable. This included steroid dependency, vascular events, white matter changes outside the radiation portal (leukoencephalopathy), hearing loss, hormonal derangements and late developing seizure disorders in the small number of long term survivors. Other altered fraction schemes have not been fruitful. Chemotherapy alone or as adjunct to radiation has demonstrated no advantage.
The present standard of care for diffuse pontine gliomas is to treat with radiation therapy alone. The volume to be treated is the T2 MRI imaging to identify the GTV. FLAIR is also used. A 1 to 1.5 cm margin is added to the GTV to construct the CTV. This is treated to a total dose of 54 Gy at 1.8 Gy/fraction over 6 weeks, which should be started promptly due to the rapid onset of neurlogic symptoms. Clinical status generall improves in 2 to 3 weeks into treatment. At this point, if steroids have been prescribed, they may be safely tapered. Improvement is impressive, but alas, short lived. Treatment at the time of progression is generally best supportive care and hospice.